Cases
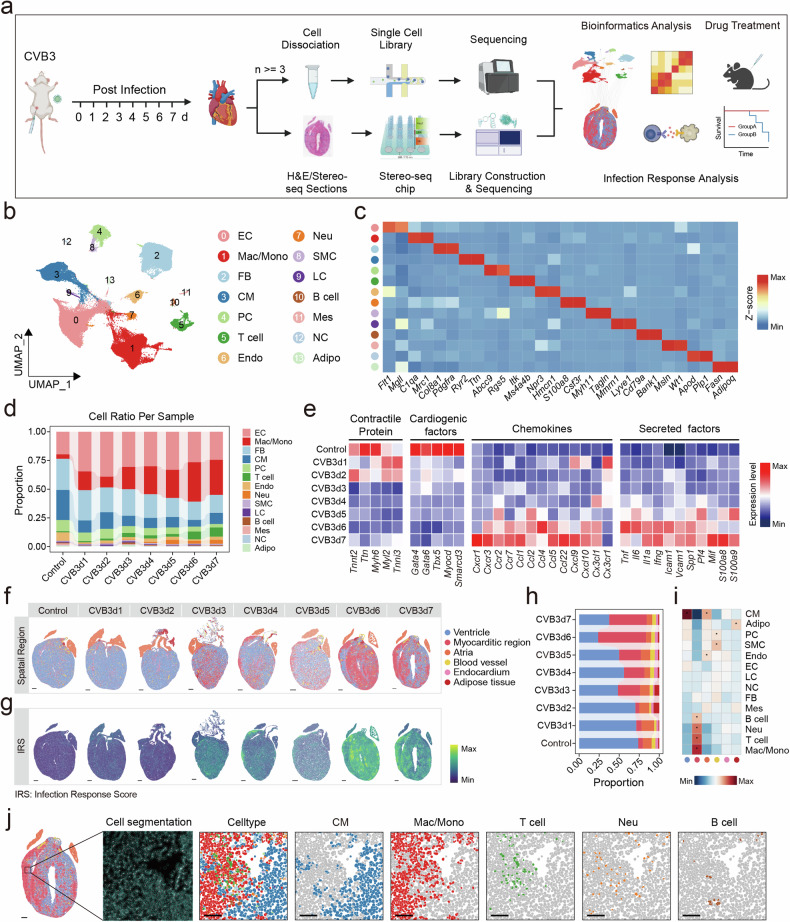
Spatiotemporal transcriptomics elucidates thepathogenesis of fulminant viral myocarditis
Fulminant myocarditis (FM) is a severe inflammatory condition of the myocardium that often results in sudden death, particularly in young individuals. In this study, we employed single-nucleus and spatial transcriptomics to perform a comprehensive analysis of coxsackievirus B3 (CVB3)-induced FM in A/J mice, spanning seven distinct time points pre- and post-treatment. Our findings reveal that mesothelial cells play a critical role in the early stage of myocarditis by acting as primary targets for CVB3 infection. This triggers the activation of macrophages, initiating a cascade of inflammation. Subsequently, pro-inflammatory Inflammatory_Mac and T cells infiltrate the myocardium, driving tissue damage. We also identified Cd8+ effector T cells as key mediators of cardiomyocyte injury. These cells release cytotoxic molecules, particularly IFN-γ, which modulates the expression of Spi1, a factor implicated in exacerbating cardiomyocyte death and amplifying disease progression. Therapeutic interventions targeting the IFN-γ/Spi1 axis demonstrated significant efficacy in FM models. Notably, intravenous immunoglobulin (IVIG) treatment reduced mortality, suppressed viral proliferation, and mitigated the hyperinflammatory state of FM. IVIG therapy also downregulated IFN-γ and Spi1 expression, underscoring its immunomodulatory and therapeutic potential. This comprehensive spatiotemporal transcriptomic analysis provides profound insights into the pathogenesis of FM and highlights actionable therapeutic targets, paving the way for more effective management strategies for this life-threatening condition.
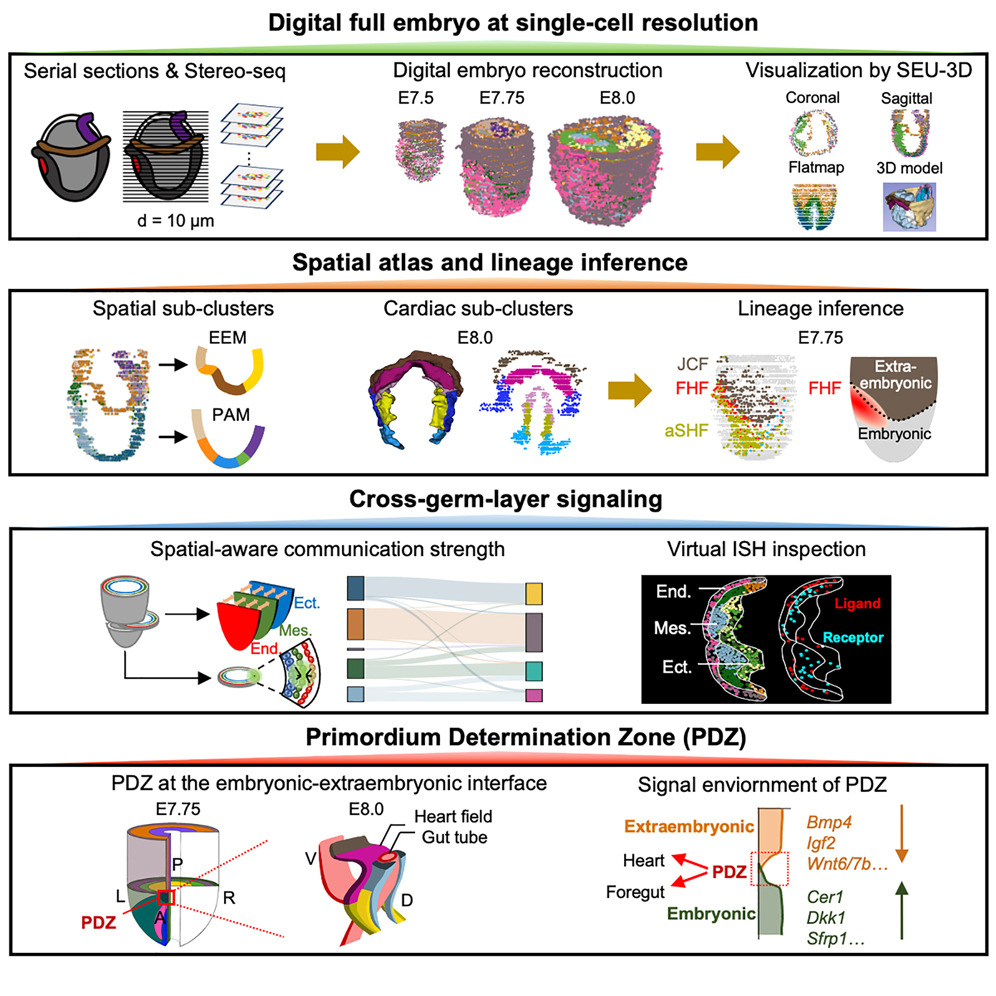
Digital reconstruction of full embryos during earlymouse organogenesis
Early organogenesis is a crucial stage in embryonic development, characterized by extensive cell fate specification to initiate organ formation but also by a high susceptibility to developmental defects. Here, we profiled 285 serial sections from six E7.5-E8.0 embryos to generate full spatiotemporal transcriptome and signal maps during early organogenesis at single-cell resolution. By developing SEU-3D, we reconstructed digital embryos, enabling investigation of regionalized gene expression in the native spatial context. We established a space-informed gene-cell co-embedding approach, systematically characterized the spatial atlas of endoderm and mesoderm derivatives, and elucidated signaling networks across germ layers and cell types. Furthermore, we characterized a primordium determination zone (PDZ) formed along the anterior embryonic-extraembryonic interface at E7.75, and it revealed that the coordinated signaling communications contribute to the formation of cardiac primordium. Collectively, the high-resolution "digital embryo" provides significant insights into early organogenesis and a unique spatial platform for studying development and diseases.

Genome sequencing reveals insights into physiology and longevity of the naked mole rat
The naked mole rat (Heterocephalus glaber) is a strictly subterranean, extraordinarily long-lived eusocial mammal. Although it is the size of a mouse, its maximum lifespan exceeds 30 years, making this animal the longest-living rodent. Naked mole rats show negligible senescence, no age-related increase in mortality, and high fecundity until death. In addition to delayed ageing, they are resistant to both spontaneous cancer and experimentally induced tumorigenesis. Naked mole rats pose a challenge to the theories that link ageing, cancer and redox homeostasis. Although characterized by significant oxidative stress, the naked mole rat proteome does not show age-related susceptibility to oxidative damage or increased ubiquitination. Naked mole rats naturally reside in large colonies with a single breeding female, the 'queen', who suppresses the sexual maturity of her subordinates. They also live in full darkness, at low oxygen and high carbon dioxide concentrations, and are unable to sustain thermogenesis nor feel certain types of pain. Here we report the sequencing and analysis of the naked mole rat genome, which reveals unique genome features and molecular adaptations consistent with cancer resistance, poikilothermy, hairlessness and insensitivity to low oxygen, and altered visual function, circadian rythms and taste sensing. This information provides insights into the naked mole rat's exceptional longevity and ability to live in hostile conditions, in the dark and at low oxygen. The extreme traits of the naked mole rat, together with the reported genome and transcriptome information, offer opportunities for understanding ageing and advancing other areas of biological and biomedical research.

The oyster genome reveals stress adaptation and complexity of shell formation
The Pacific oyster Crassostrea gigas belongs to one of the most species-rich but genomically poorly explored phyla, the Mollusca. Here we report the sequencing and assembly of the oyster genome using short reads and a fosmid-pooling strategy, along with transcriptomes of development and stress response and the proteome of the shell. The oyster genome is highly polymorphic and rich in repetitive sequences, with some transposable elements still actively shaping variation. Transcriptome studies reveal an extensive set of genes responding to environmental stress. The expansion of genes coding for heat shock protein 70 and inhibitors of apoptosis is probably central to the oyster's adaptation to sessile life in the highly stressful intertidal zone. Our analyses also show that shell formation in molluscs is more complex than currently understood and involves extensive participation of cells and their exosomes. The oyster genome sequence fills a void in our understanding of the Lophotrochozoa.
Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator
The organized societies of ants include short-lived worker castes displaying specialized behavior and morphology and long-lived queens dedicated to reproduction. We sequenced and compared the genomes of two socially divergent ant species: Camponotus floridanus and Harpegnathos saltator. Both genomes contained high amounts of CpG, despite the presence of DNA methylation, which in non-Hymenoptera correlates with CpG depletion. Comparison of gene expression in different castes identified up-regulation of telomerase and sirtuin deacetylases in longer-lived H. saltator reproductives, caste-specific expression of microRNAs and SMYD histone methyltransferases, and differential regulation of genes implicated in neuronal function and chemical communication. Our findings provide clues on the molecular differences between castes in these two ants and establish a new experimental model to study epigenetics in aging and behavior.
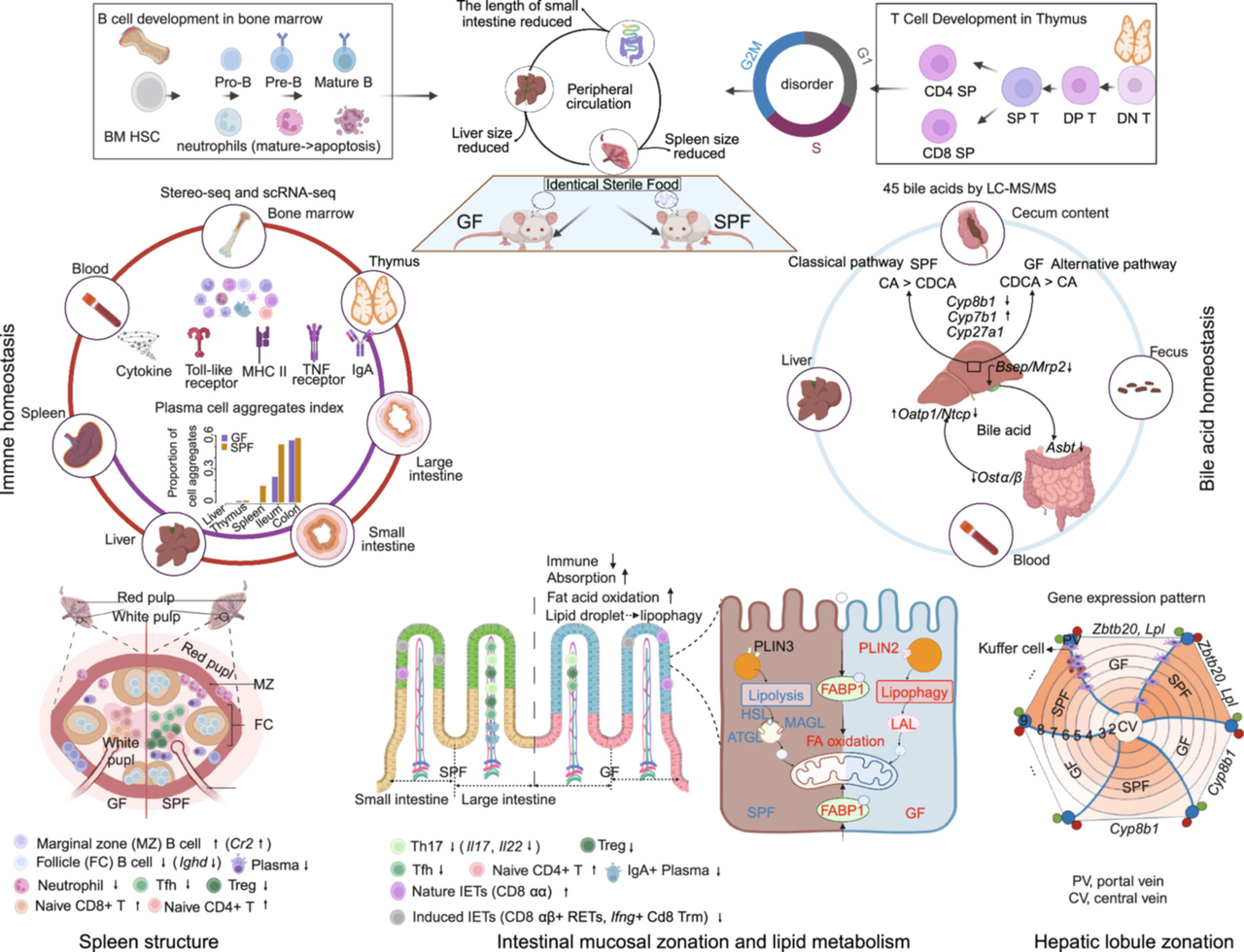
Cross-tissue multi-omics analyses reveal the gut microbiota's absence impacts organ morphology, immune homeostasis, bile acid and lipid metabolism
The gut microbiota influences host immunity and metabolism, and changes in its composition and function have been implicated in several non-communicable diseases. Here, comparing germ-free (GF) and specific pathogen-free (SPF) mice using spatial transcriptomics, single-cell RNA sequencing, and targeted bile acid metabolomics across multiple organs, we systematically assessed how the gut microbiota's absence affected organ morphology, immune homeostasis, bile acid, and lipid metabolism. Through integrated analysis, we detect marked aberration in B, myeloid, and T/natural killer cells, altered mucosal zonation and nutrient uptake, and significant shifts in bile acid profiles in feces, liver, and circulation, with the alternate synthesis pathway predominant in GF mice and pronounced changes in bile acid enterohepatic circulation. Particularly, autophagy-driven lipid droplet breakdown in ileum epithelium and the liver's zinc finger and BTB domain-containing protein (ZBTB20)-Lipoprotein lipase (LPL) (ZBTB20-LPL) axis are key to plasma lipid homeostasis in GF mice. Our results unveil the complexity of microbiota-host interactions in the crosstalk between commensal gut bacteria and the host.

Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing
Domestic pigs have evolved genetic adaptations to their local environmental conditions, such as cold and hot climates. We sequenced the genomes of 69 pigs from 15 geographically divergent locations in China and detected 41 million variants, of which 21 million were absent from the dbSNP database. In a genome-wide scan, we identified a set of loci that likely have a role in regional adaptations to high- and low-latitude environments within China. Intriguingly, we found an exceptionally large (14-Mb) region with a low recombination rate on the X chromosome that appears to have two distinct haplotypes in the high- and low-latitude populations, possibly underlying their adaptation to cold and hot environments, respectively. Surprisingly, the adaptive sweep in the high-latitude regions has acted on DNA that might have been introgressed from an extinct Sus species. Our findings provide new insights into the evolutionary history of pigs and the role of introgression in adaptation.
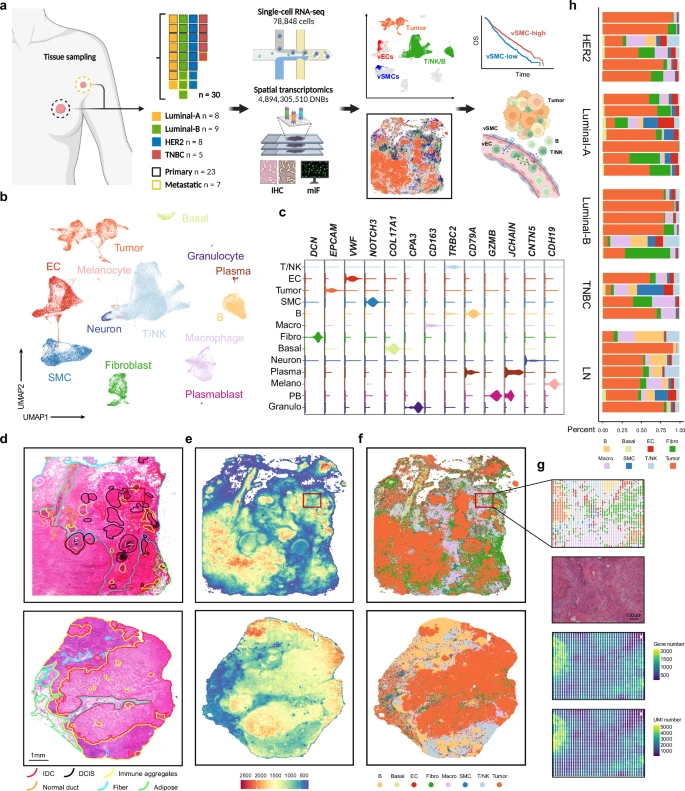
Spatially resolved atlas of breast cancer uncovers intercellular machinery of venular niche governing lymphocyte extravasation
Breast cancers present intricate microenvironments comprising heterotypic cellular interactions, yet a comprehensive spatial map remained to be established. Here, we employed the DNA nanoball-based genome-wide in situ sequencing (Stereo-seq) to visualize the geospatial architecture of 30 primary breast tumors and metastatic lymph nodes across different molecular subtypes. This unprecedented high-resolution atlas unveils the fine structure of the tumor vasculature, highlighting heterogeneity in phenotype, spatial distribution, and intercellular communication within both endothelial and perivascular cells. In particular, venular smooth muscle cells are identified as the primary source of CCL21/CCL19 within the microenvironment. In collaboration with ACKR1-positive endothelial cells, they create a chemokine-rich venular niche to synergistically promote lymphocyte extravasation into tumors. High venule density predicts increased immune infiltration and improved clinical outcomes. This study provides a detailed spatial landscape of human breast cancer, offering key insights into the venular regulation of tumor immune infiltration.
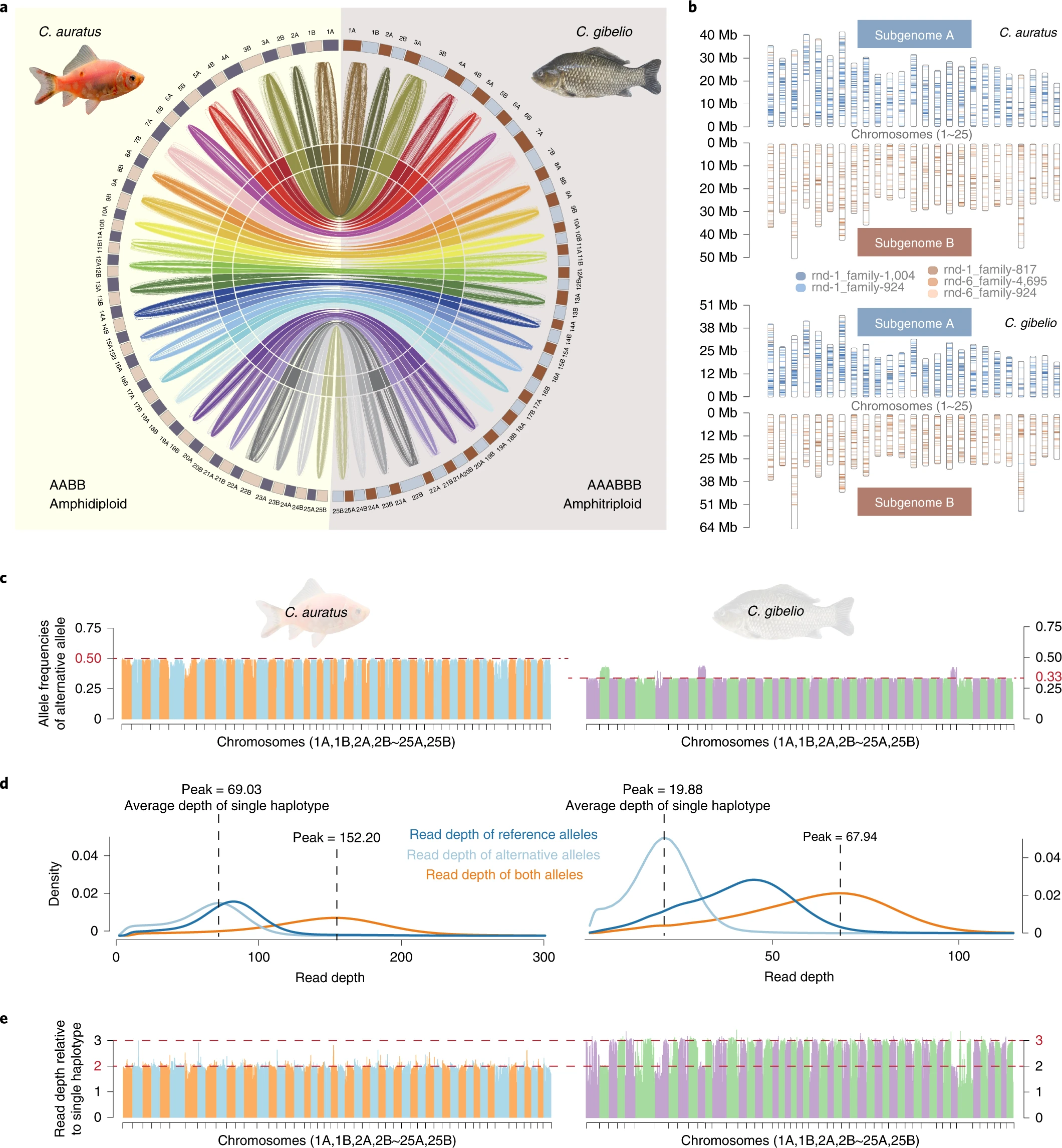
Comparative genome anatomy reveals evolutionary insights into a unique amphitriploid fish
Triploids are rare in nature because of difficulties in meiotic and gametogenic processes, especially in vertebrates. The Carassius complex of cyprinid teleosts contains sexual tetraploid crucian carp/goldfish (C. auratus) and unisexual hexaploid gibel carp/Prussian carp (C. gibelio) lineages, providing a valuable model for studying the evolution and maintenance mechanism of unisexual polyploids in vertebrates. Here we sequence the genomes of the two species and assemble their haplotypes, which contain two subgenomes (A and B), to the chromosome level. Sequencing coverage analysis reveals that C. gibelio is an amphitriploid (AAABBB) with two triploid sets of chromosomes; each set is derived from a different ancestor. Resequencing data from different strains of C. gibelio show that unisexual reproduction has been maintained for over 0.82 million years. Comparative genomics show intensive expansion and alterations of meiotic cell cycle-related genes and an oocyte-specific histone variant. Cytological assays indicate that C. gibelio produces unreduced oocytes by an alternative ameiotic pathway; however, sporadic homologous recombination and a high rate of gene conversion also exist in C. gibelio. These genomic changes might have facilitated purging deleterious mutations and maintaining genome stability in this unisexual amphitriploid fish. Overall, the current results provide novel insights into the evolutionary mechanisms of the reproductive success in unisexual polyploid vertebrates.
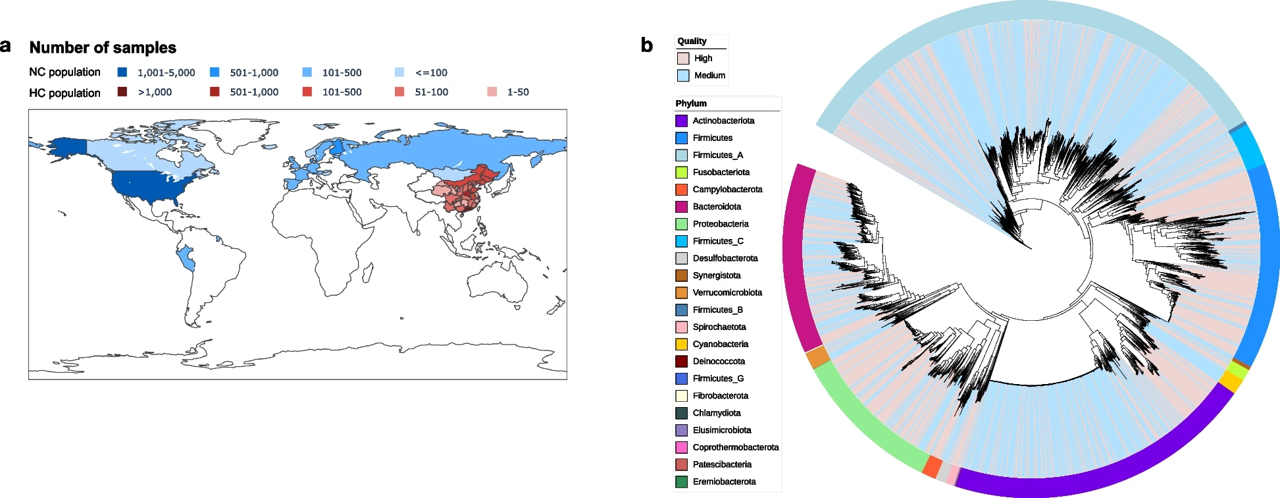
Exploring differences in the human gut microbiome between Han Chinese and non-Chinese populations
Background: The human gut microbiota exhibits significant diversity across populations, influenced by factors such as geography, diet, and lifestyle, particularly between the Han Chinese and non-Chinese populations. While previous studies have predominantly focused on the taxonomic abundance of the gut microbiome, the impact of single nucleotide polymorphisms (SNPs) in driving population-specific differences remains largely underexplored. Results: In this study, we systematically investigated gut microbial differences between the Han Chinese and non-Chinese populations using the Human Gut Microbiome Reference Genome Catalog (HGMRGC). We observed geography was the primary driver of microbial variation of abundance and SNPs. We identified 689 population-specific genome clusters from the Collinsella genus with functional differences in carbohydrate utilization and 108 species exhibiting distinct prevalence related to vitamin biosynthesis, antibiotic resistance, and carbohydrate metabolism. Beta diversity analysis highlighted significant inter-population differences in both microbial abundance and SNPs, while alpha diversity analysis revealed that non-Chinese populations exhibited higher diversity in microbial abundance, and Han Chinese populations displayed greater diversity in SNPs. Conclusions: This study offers a comprehensive analysis of gut microbial differences between Han Chinese and non-Chinese populations, highlighting the profound influence of population-specific traits on microbial diversity and function. We also provide a comprehensive human gut microbial reference genome catalog, with a particular focus on the Han Chinese population, laying a foundation for future research on gut microbiota genomic variations.